Our laboratory unravels mechanisms of virus biology, drug action and drug resistance. These basic science studies are fundamentally important as prerequisites for the development of novel treatments of serious diseases.
Significance:
Despite breakthroughs in the treatment of viral diseases, they remain enormous public health challenges. Over 30 million people are currently infected with Human Immunodeficiency Virus (HIV). Remarkably, despite the availability of a vaccine for Hepatitis B Virus (HBV), ~400 million individuals are chronically infected and in need of treatment with antivirals. In this era of globalization, worldwide outbreaks of new, emerging, and re-emerging diseases including Middle East Respiratory Syndrome (MERS) and Severe Acute Respiratory Syndrome (SARS), or Foot-and-Mouth-Disease (FMD) are global threats that have the potential to become an immense burden on the US and world economies. Recently, we have been living a pandemic, COVID-19, that is caused by coronavirus SARS-CoV-2, and that has resulted in millions of deaths throughout the globe. Our laboratory is working hard on contributing to the understanding of coronavirus biology, developing tools and helping develop therapeutics against SARS-CoV-2.
Academic research is the workhorse of drug discovery, as it focuses on fundamental virus biology and molecular interactions that involve viral targets, potential therapeutics, drug resistant proteins, and host factors. In depth understanding of these interactions is needed for innovative and novel drug discoveries and this is an area where we have made significant contributions. Toward that end we use a combination of crystallographic, biochemical, virological approaches to chart the details of molecular recognition during steps of the life cycle of viruses, allowing us to identify and target weak spots.
- Design and Synthesis of New GS-6207 Subtypes for Targeting HIV-1 Capsid Protein
- A Tag-Free Platform for Synthesis and Screening of Cyclic Peptide Libraries
- Identification of clickable HIV-1 capsid-targeting probes for viral replication inhibition
- Synthesis and biological evaluation of novel peptidomimetic inhibitors of the coronavirus 3C-like protease
- Use of TSAR, Thermal Shift Analysis in R, to identify Folic Acid as a Molecule that Interacts with HIV-1 Capsid
- HIV-2 inhibits HIV-1 gene expression via two independent mechanisms during cellular co-infection
- Discovery of Nirmatrelvir Resistance Mutations in SARS-CoV-2 3CLpro: A Computational-Experimental Approach
- HIV-1 Resistance to Islatravir/Tenofovir Combination Therapy in Wild-Type or NRTI-Resistant Strains of Diverse HIV-1 Subtypes
- Mechanisms by which the cystic fibrosis transmembrane conductance regulator may influence SARS-CoV-2 infection and COVID-19 disease severity
- Multidisciplinary studies with mutated HIV-1 capsid proteins reveal structural mechanisms of lattice stabilization
- Biology of the hepatitis B virus (HBV) core and capsid assembly modulators (CAMs) for chronic hepatitis B (CHB) cure
- The HIV-1 capsid core is an opportunistic nuclear import receptor
- Targeting the HIV-1 and HBV Capsids, an EnCore
- Mechanisms of Action of the Host-Targeting Agent Cyclosporin A and Direct-Acting Antiviral Agents against Hepatitis C Virus
- TMPRSS2 and SARS-CoV-2 SPIKE interaction assay for uHTS

SARS-CoV-2 replicon and virus-like particles
Innovative virological tools are essential to study the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication cycle and pursue drug development under routine biocontainment. Stable cell lines (including human) harboring replicons were prepared and multiplex high-throughput assays using orthogonal reporters were established. Replicons containing active site mutations, drug resistant mutations, and genotypically diverse genes (representing alpha, beta, delta, or omicron variants) were employed in mechanistic studies. Production of single-round infectious virus-like particles (VLPs) by replicon co-expression with structural proteins enabled virus assembly and entry studies at biosafety level 2 (BSL2) facilities. For proof-of-concept, we generated VLPs carrying replicon lacking membrane (M) protein in the genome, which were trans-encapsidated with M during particle assembly. These VLPs were capable of multi-round infections when M was present in susceptible target cells. Using infectious VLPs with open reading frame (ORF) deletions we showed that the loss of ORFs 6, 7, and 8 produced a moderate reduction in infectious titer, whereas the additional loss of ORF3 reduced the titer by 80%. These strategies enable the use of BSL2 facilities for studies addressing drug discovery, mechanisms of inhibition and resistance, and for characterizing the role of viral genes and host dependency factors. The high flexibility of the VLP system enables facile assembly of chimeric VLPs, which, unlike pseudotype-based systems, form authentic SARS-CoV-2 virions and could be expeditiously used for testing vaccine efficacy, susceptibility to antibodies, structural determination of virions, and vaccine development studies.
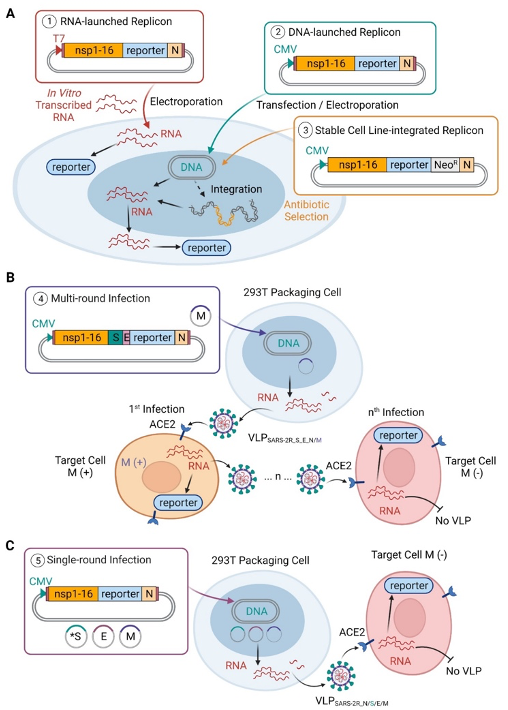
Five strategies for using SARS-2R replicon systems
(A) SARS-2R constructs (details in Table S1) containing reporter gene(s) with or without antibiotic resistance genes are applicable for research at BSL2 level containment. RNA-launched replicon is driven by T7 promoter and requires in vitro transcription prior to electroporation of viral RNA into cells (①). DNA-launched replicon driven by CMV promoter allows direct transfection or electroporation of DNA into cells (②). By including neomycin resistance gene in the construct, stable cell lines with integrated SARS-2R can be selected with geneticin (G418), an analog of neomycin sulfate (③).
(B) Replicon-derived VLPs can be generated for multi-round infections (④). VLPSARS-2R_S_E_N/M is generated by transfection of two plasmids, pBAC-SARS-2R_SE_mNG_N, and M expression plasmid in 293T packaging cells. In M (+) cells, presence of M allows production of VLPSARS-2R_S_E_N/M for next round of infection. This process may continue for multiple rounds (denoted by n) with the presence of M for assembly of VLP. In M (-) cells, absence of M prevents production of VLPs and subsequent round of infection stops.
Nipah virus minigenome
Nipah virus (NiV) is a highly dangerous zoonotic pathogen with alarmingly high case fatality rates, ranging from 71% to nearly 100%. Currently, there are no approved treatments or vaccines for NiV, highlighting the need for effective therapeutics. By leveraging minigenome (MG) replicon-based approaches, we aim to identify compounds that demonstrate potent antiviral activity against NiV, which can serve as starting points for the development of novel antiviral therapies.
Towards that end, we have generated a NiV MG replication system using nano luciferase and mNeonGreen orthogonal reporters. Through careful adjustments of the MG construct and the expression of essential proteins, we have achieved a high signal-to-background level suitable for high-throughput screening (HTS). However, as the MG system involves transfection of multiple plasmids, it is important that we improve the efficiency of the system and expand its use in diverse target cells. To address this, we plan to consolidate several of the viral proteins and reporter genes into a single plasmid for multiple protein expression, use a lentivirus system for delivery, test the MG in various cell lines and primary cells, and establish MG-expressing stable cell lines. These steps aim to enhance transfection efficiency and make the MG system more versatile and reliable for rigorous screening.
In our preliminary data, we have been using our first generation of the MG to screen a unique nucleoside analog library that collaborator Schinazi and colleagues, including co-I Amblard, have built over the course of decades. Initial experiments on a small portion of the library have already yielded promising results, identifying a potent nucleoside analog hit with an EC50 of 2.6 µM in HEK293T cells. Further screening of the library will be conducted, and compound hits will be confirmed by synthesizing fresh inhibitors and testing them in various cell lines and primary cells using optimized versions of the NiV MG replication system. The triphosphate forms of the nucleoside analogs will also be prepared and assessed for their ability to inhibit RNA synthesis by the NiV RNA polymerase. Compounds will be prioritized on the basis of potency and cytotoxicity. Future goals include validation using infectious NiV (BSL-4). These endeavors will lay the foundation for future chemical optimization of the most promising inhibitors, aiming to enhance their potency, cytotoxicity, ADME (absorption, distribution, metabolism, and excretion), PK (pharmacokinetic) properties, and their efficacy in animal models, towards the discovery of effective antiviral drugs to combat NiV infection.
A) All four components (MG, N, L, P) are essential for Nipah minigenome replication, which is susceptible to RDV. B) Compound Remdesivir (RDV) susceptibility assay with Nipah minigenome system in 96-well format.
Mpox, caused by the orthopoxvirus mpox virus (MPXV), became an infectious disease of worldwide concern following a 2022 global outbreak in which more than 80,000 cases were confirmed in more than 100 countries, mostly in countries outside the endemic regions of West and Central Africa. Because cases and outbreaks of mpox are still appearing, there is a need to be prepared through the development of antiviral therapies targeting MPXV. Current therapies, comprised of tecovirimat (TPOXX), cidofovir (CDV), and brincidofovir, have concerns with toxicity and potential for emergence of drug resistance. Therefore, there are currently no mpox treatments that are both highly potent and well-tolerated.
Research done by our lab has shown that the repurposed compounds tenofovir alafenamide (TAF) and adefovir dipivoxil, which are acyclic nucleoside phosphonates, inhibit MPXV DNA replication.* However, these compounds should be tested for the potential for antiviral resistance to emerge before they could be used in animal studies and clinical trials. My current research goals are to investigate the drug resistance potential of orthopoxviruses to these phosphonates and continue screening for orthopoxvirus inhibitors.
*Lee J, Boggs EA, Zhang H, Tedbury PR, Sarafianos SG. Mpox Virus is Inhibited By Nucleoside Analogues Including the Acyclic Phosphonates Tenofovir and Adefovir. bioRxiv. 2023:2023.06.30.547277. doi: 10.1101/2023.06.30.547277
Stopping the virus in its tracks
Targeting the translocation function of HIV Reverse transcriptase (RT) with EFdA (Fig.1), the most potent anti-HIV drug
Our lab has discovered an ultrapotent mechanism of HIV inhibition. This mechanism is followed by EFdA, a compound that is by far the most potent inhibitor of HIV replication inhibiting HIV at low picomolar concentrations by an innovative mechanism of action and a very promising resistance profile (Kawamoto et al. 2008, Michailidis et al. 2009). EFdA was originally synthesized by Ohrui and recently licensed by Yamasa to Merck for the treatment of HIV infection.
Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) efficiently suppress HIV and serve as backbone of antiretroviral therapies. However, new therapeutics are needed for continued control of HIV infection. Research from our lab contributed to the preclinical development of 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) (near 30 EFdA/islatravir-related papers) and its subsequent advance in the clinic. Our work on the exceptional potency, stability, unique mechanism, and attributes of EFdA contributed to its licensing by Merck, who successfully used it in Phase I, and subsequently introduced it in Phase II (alternate EFdA names: MK-8591, Islatavir) and now phase III clinical trials. EFdA has generated “compelling early results for both treatment and prevention” in patients (http://i-base.info/htb/32246), tested at very low doses (>10,000 lower than some NRTIs) aiming at once-weekly oral dosing, once-yearly slow-release dosing, unprecedented modalities for HIV therapies.
a) We have shown that EFdA has high potency in vitro, in cell culture (EC50=50 pM in PBMCs), mice and non-human primate animal models. Although EFdA retains the 3’-OH, it blocks HIV reverse transcriptase (RT) in vitro, by acting either as an immediate or as delayed chain terminator (ICT, DCT; Fig. 2) due to translocation inhibition of EFdA-terminated viral DNA. Thus, even though EFdA is an NRTI, it is referred to as a nucleotide reverse transcriptase translocation inhibitor (NRTTI) to highlight its unique mechanism of action (MoA). b) Our recent data include paradigm-shifting NRTI-resistance findings: counter to the long-held belief that nucleoside analog drug resistance can occur through only one of two mutually exclusive mechanisms of NRTI resistance, we demonstrate that HIV mutants have modest EFdA resistance through simultaneously BOTH decreased inhibitor incorporation and enhanced inhibitor excision mechanisms

Hypersusceptibility of tenofovir-and nonnucleoside RT inhibitor-resistant viruses to EFdA: Since most HIV patients are currently on primarily tenofovir-based therapies, we assessed the effect of EFdA and its analogs on HIVs that are resistant to nucleoside RT inhibitors (NRTIs) and nonnucleoside RT inhibitors (NNRTIs). EFdA suppressed NRTI-resistant viruses with remarkable efficiency and was remarkably effective against the tenofovir-resistant variants.
The mechanism of this hypersensitivity was shown to involve suppression of the mild excision activity of RT that result in mold unblocking of EFdA-terminated priomers in WT, but not in K65R mutants (Fig. 3).
This is highly important because tenofavir is the first line of defense in HIV therapies and a backup drug that works well with tenofavir-resistant viruses would be very valuable. Our recently published relevant manuscript (Michailidis et al. 2013)is among the highest articles ever scored in Retrovirology (a journal ranked #1 in Virology and #16 of 452 in the broader field) and in the top 5% of all of the 2.128,264 articles tracked by Altmetric across all journals. In another recent manuscript (Hachiya et al. 2013) we showed that EFdA acts synergistically with the second generation NNRTI rilpivirine and does not compete with tenofovir (Fig.4). Collectively, these data predict that EFdA would form a formidable combination with tenofovir, and/or rilpivirine and would perform extremely well even with patients that have failed current therapies.


Our crystallographic work (10.1073/pnas.1605223113) has shown that the structural attributes of EFdA impact interactions with diverse RTs (wild-type, drug-resistant, viruses) leading to clinically important differences in efficiency of inhibition, resistance, hypersusceptibility, and biochemical MoA (Fig. 5).

Extraordinary in vivo activity of EFdA: Importantly, our collaborative non-human primate study with Drs. M. Murphey-Corb, Michael Parniak at the University of Pittsburgh and Hiroaki Mitsuya (NIH, University of Kumamoto) established that EFdA has extraordinary activity in vivo (Fig.6). Moreover, no detectable clinical or pathological signs of drug toxicity were observed within 6 months of continuous therapy. Following our exciting data on EFdA, Merck licensed EFdA for preclinical development. Overall, our EFdA-related work (so far 14 relevant publications) has been instrumental for the advancement of EFdA and it has been carried out in collaboration with Drs. Hiroaki Mitsuya (NIH, Kumamoto University), Michael Parniak (University of Pittsburgh), Eiichi Kodama (Tokoku University), and recently Lesa Rohan (University of Pittsburgh). It has been funded by NIH (R01AI076119, R21/R33AI079801, R01AI076119-S2, R01AI076119-06AI) and amfAR grants.

Project Title: Ultrapotent Inhibitors of Wild-type and Multi-drug Resistant HIV
Project PI: Sarafianos, Stefan G (Contact PI)
Project Number: R37AI076119
Project Description: Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) efficiently suppress HIV and serve as backbone of antiretroviral therapies. However, new therapeutics are needed for continued control of HIV infection. 4’- Ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) is an NRTI with exceptional potency, stability, and unique mechanism of action against HIV, which has been licensed by Merck. EFdA (also known as MK-8591 or Islatavir) has been successfully used in Phase I and recently introduced in Phase II clinical trials. EFdA has generated “compelling early results for both treatment and prevention” in patients, tested at remarkably low doses (up to >10,000-fold lower doses than current NRTI drugs) aiming at once-weekly oral and once-yearly slow-release dosing, unprecedented modalities for HIV therapies. Hence, this work is directly relevant to NIH guidelines for high priority AIDS funding (NOT-OD-15-137) for “next generation therapies…that are longer acting.” We have shown that a) EFdA has high potency in vitro, in cell culture (EC50=50 pM in PBMCs), mice and non- human primate animal models. Although EFdA retains the 3’-OH, it blocks HIV reverse transcriptase (RT) in vitro, primarily as an immediate/obligate and at times delayed chain terminator due to difficulty of EFdA- terminated viral DNA to translocate. Thus, EFdA is termed a nucleotide reverse transcriptase translocation inhibitor (NRTTI). However, the inhibition mechanism of EFdA in HIV-infected cells (primary or cell lines) remains unknown. Toward that end, Co-I Malim has established an innovative assay that enables study of the EFdA antiviral mechanism in HIV-infected cells. Additionally, in vitro passage experiments have identified mutations that impart EFdA resistance through two paradigm-shifting dual mechanisms or resistance: decreased incorporation/enhanced excision and decreased incorporation/enhanced translocation. Data on an EFdA analog suggest efficient inhibition of EFdA-resistant HIV, although the mechanism of this phenomenon is not understood. Notably, treatment with key new generation NNRTI doravirine (DOR) leads to resistance mutations that impart hypersusceptibility to EFdA, paving the way for EFdA/DOR combination therapies. We hypothesize that the structural attributes of EFdA and its analogs impact interactions with diverse RTs (wild-type, drug- resistant, from diverse clades, viruses), leading to clinically important differences in efficiency of inhibition, resistance, hypersusceptibility, and biochemical mechanism of action. This hypothesis will be tested through three specific aims, which endeavor to answer the above questions using a combination of novel sequencing, biochemical, structural, and virological approaches. This work will help optimize combination therapies that reduce drug burden and have exceptional long-acting potential, addressing adherence challenges of chronic HIV treatment.
Some references relevant to our EFdA-related work:
https://projectreporter.nih.gov/project_info_results.cfm?aid=9459305&icde=45906984
Project Title: Novel mechanism of integrase (IN) resistance to Dolutegravir through epistatic interactions between IN and the nucleocapsid and polypurine tract regions of HIV-1
Project PI: Sarafianos, Stefan G (Contact PI); Hachiya, Atsuko (MPI); Lyumkis, Dmitry (MPI)
Project Number: 5R01AI146017
Project Description: Integration is essential for HIV-1 replication and is completed by integrase (IN). A class of drugs which inhibit the strand transfer (ST) function of HIV integrase, called IN strand transfer inhibitors (INSTIs), includes approved drugs raltegravir (RAL), elvitegravir (EVG) (1st generation) and dolutegravir (DTG), bictegravir (BIC) (2nd generation). DTG has a higher genetic barrier to resistance than RAL or EVG, and is recommended by the World Health Organization as an alternative to efavirenz in first-line regimens in low- and middle-income countries (LMICs). Selection for DTG resistance is rare, but does exist and is currently not well understood. There is mounting evidence for failure of DTG-based treatment in clinical trials (VIKING-3 study) in the absence of mutations in the targeted IN gene. Our overarching hypothesis is that mutations outside IN can impart drug resistance to IN-targeting drugs through indirect interactions that we call epistatic. The scientific premise for studying these interactions is soundly grounded on two key pieces of evidence: First, in surprising preliminary data from in vitro serial passage experiments in the presence of increasing amounts of DTG, a DTG resistance mutation located outside IN was discovered. Experiments with recombinant viruses validated DTG resistance of this mutation and showed enhanced resistance in the presence of the E157Q IN polymorphism. Moreover, deep sequencing analyses showed that compared to infection by wild-type, mutant–containing viruses resulted in more insertions, deletions, and non-canonical long terminal repeat (LTR) ends in 2-LTR circles and integrated viral DNA. Second, a recent independent study based on similar serial passage experiments, identified changes at the general G-tract region of the 3’-polypurine tract (3’-PPT) in a DTG-resistant virus (Malet et al., 2017). Subsequently, different 3’-PPT changes were reported in a patient that failed DTG therapy. However, the mechanism of DTG resistance through mutations at the 3’-PPT remains unclear due to conflicting hypotheses and lack of experimental validation. Our hypothesis is that mutations outside IN can affect DTG resistance by altering the LTR ends at the INSTI binding site. This hypothesis will be tested by a team of experts that includes PIs Sarafianos (biochemical, virological drug resistance mechanisms), PI Lyumkis (single particle cryo-EM on intasome/drug complexes) and PI Hachiya (virology, drug resistance) with the support by HIV IN expert Hughes (NCI), using virological, biochemical, and structural tools to address the aims to investigate the virological, biological, and structural mechanisms of DTG resistance through epistatic interactions. These innovative studies will help elucidate the molecular mechanisms of INSTI resistance through epistatic interactions via mutations that are outside the IN gene and affect the INSTI-binding site. They are significant and will help explain clinical failures to DTG-based regimens in the absence of mutations in IN
Publications (selected) citing the Project:
1. “Structural basis for strand-transfer inhibitor binding to HIV intasomes.” Passos DO, Li M, Jóźwik IK, Zhao XZ, Santos-Martins D, Yang R, Smith SJ, Jeon Y, Forli S, Hughes SH, Burke TR Jr, Craigie R, Lyumkis D. Science. 2020 367(6479):810-814.

2. “HIV-1 Integrase Inhibitors That Are Active against Drug-Resistant Integrase Mutants.”Smith, Steven J; Zhao, Xue Zhi; Passos, Dario Oliveira; Lyumkis, Dmitry; Burke Jr, Terrence R; Hughes, Stephen H. Antimicrobial agents and chemotherapy 2020 08 20; 64 (9).

3. ”Single-cell Multiplexed Fluorescence Imaging to Visualize Viral Nucleic Acids and Proteins and Monitor HIV, HTLV, HBV, HCV, Zika Virus, and Influenza Infection.” Shah, Raven; Lan, Shuiyun; Puray-Chavez, Maritza N; Liu, Dandan; Tedbury, Philip R; Sarafianos, Stefan G. Journal of visualized experiments: JoVE 2020 10 29; (164)

4. “Structural Biology of HIV Integrase Strand Transfer Inhibitors.” Jóźwik, Ilona K; Passos, Dario O; Lyumkis, Dmitry. Trends in pharmacological sciences 2020 09; 41 (9) 611-626.

5. “The SMC5/6 complex compacts and silences unintegrated HIV-1 DNA and is antagonized by Vpr.” Dupont, Liane; Bloor, Stuart; Williamson, James C; Cuesta, Sergio Martínez; Shah, Raven; Teixeira-Silva, Ana; Naamati, Adi; Greenwood, Edward J D; Sarafianos, Stefan G; Matheson, Nicholas J; Lehner, Paul J Cell host & microbe 2021 05 12; 29 (5) 792-805.e6

6. “Specific mutations in the HIV-1 G-tract of the 3’-Polypurine Tract cause resistance to integrase strand transfer inhibitors” Author(s): Hachiya, Atsuko; Kuboya, Mai; Urara, Shigemi; Ode, Hirotaka; Yokomaku, Yoshiyuki; Kirby, Karen; Sarafianos, Stefan; Iwatani, Yasumasa. Journal of Antimicrobial Therapeutics, 2021 in press.

Targeting HIV RNase H, the ‘final frontier’
CA is a structural protein essential for early and late events of HIV replication. Its multiple roles in infection and pathogenesis are not entirely understood. CA is an attractive therapeutic target since proper capsid formation is required for virus infectivity. We have discovered 18E8 (unpublished, to be patented), a compound with antiviral properties that interferes with the rate of CA multimerization. We have shown 18E8 to work by a novel mechanism and recently received an NIH grant to study this mechanism. Importantly, during this study we have solved the crystal structure of native hexameric full length HIV CA, an elusive structure for ~20 years. The structure provides insights into HIV biology and has resulted into collaborations with distinguished scientists in the field. Future efforts focus on providing the molecular details of CA interactions with cellular host proteins and improving our early inhibitors towards the development of innovative CA-targeting HIV therapeutics.
Discovery and characterization of a new mechanism of HIV multi-drug resistance
In collaboration with Japanese clinical scientists and independently from competing groups we helped demonstrate that RT mutation N348I causes resistance to drugs that belong to multiple classes of antiretrovirals (PI: Eiichi Kodama) (Hachiya et al. 2008). Our findings contributed to the redesign of clinical tools that detect HIV resistance mutations in patients and define the course of therapies. We also discovered new RT mutati on K70Q that causes multidrug resistance to tenofovir-based regimens (Hachiya et al, 2011). Ongoing and future efforts focus on the study of polymorphisms in various HIV clades towards elucidating drug resistance differences among various HIV subtypes, obtaining knowledge that should help optimize treatment strategies.
Novel methods for identification of drug resistance in HIV and HBV patients
In a related effort on drug resistance, we accepted an invitation for collaboration by Genematrix, a publicly traded Korean company, which manufactures mass spectrometry instrumentation. Based on our experience with drug resistance in HIV and other viruses Genematrix and ourgroup published a method that increases the sensitivity of current drug-resistance detection protocols, so that we can detect earlier drug resistance mutations and accordingly direct changes in therapeutic regimens (funding by 5-yr grant from the Korean government/Genematrix).
Discovery of FMDV inhibitors
FMDV is a picornavirus that infects cloven-hoofed animals and leads to severe losses in livestock. We discovered and characterized a novel inhibitor of FMDV replication. Through collaboration with USDA scientists at Plum Island NY (Dr. E. Rieder and colleagues) we demonstrated antiviral activity of the compound (Durk et al, 2010), and now have an international patent for its use. We have shown that the novel targeted pocket is conserved in other viruses which we are currently studying.
Discovery of multiple assays for optimal combinations of HCV inhibitors
We have recently started a project on HCV in collaboration with world-leader in the field Dr. Charles Rice (Rockefeller University) and obtained 5-yr NIH funding R01AI099284. We have established novel microscopy-based assays for identifying optimal combinations of anti-HCV drugs. We are using these novel technologies to address the effect of host factors in the HCV life cycle. These studies were initiated by research faculty in our lab Dr. R. Ralston, who is now a Research Professor at Kansas Univ. Medical Center.